
Model card

AlphaFold
This package provides an implementation of the inference pipeline of AlphaFold
v2. For simplicity, we refer to this model as AlphaFold throughout the rest of
this document.
We also provide:
- An implementation of AlphaFold-Multimer. This represents a work in progress
and AlphaFold-Multimer isn't expected to be as stable as our monomer
AlphaFold system. Read the guide for how
to upgrade and update code. - The technical note containing the models
and inference procedure for an updated AlphaFold v2.3.0. - A CASP15 baseline set of predictions along
with documentation of any manual interventions performed.
Any publication that discloses findings arising from using this source code or
the model parameters should cite the
AlphaFold paper and, if
applicable, the
AlphaFold-Multimer paper.
Please also refer to the
Supplementary Information
for a detailed description of the method.
You can use a slightly simplified version of AlphaFold with
this Colab notebook
or community-supported versions (see below).
If you have any questions, please contact the AlphaFold team at
alphafold@deepmind.com.
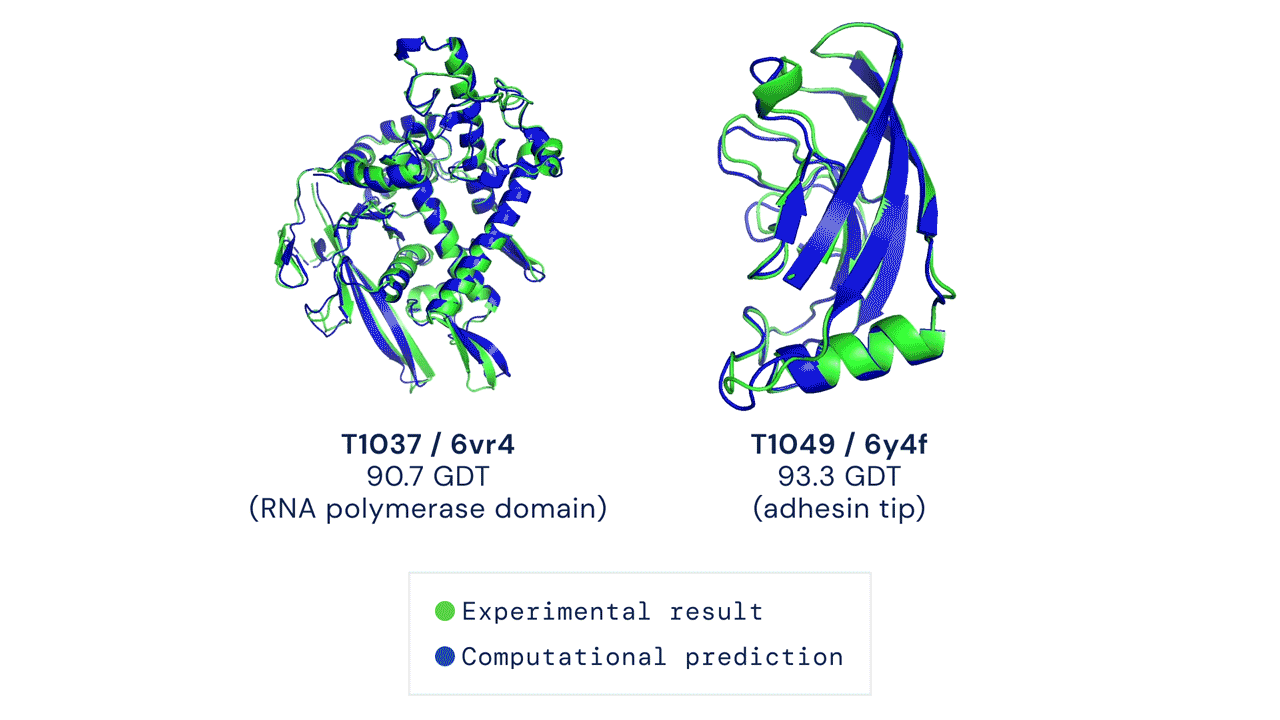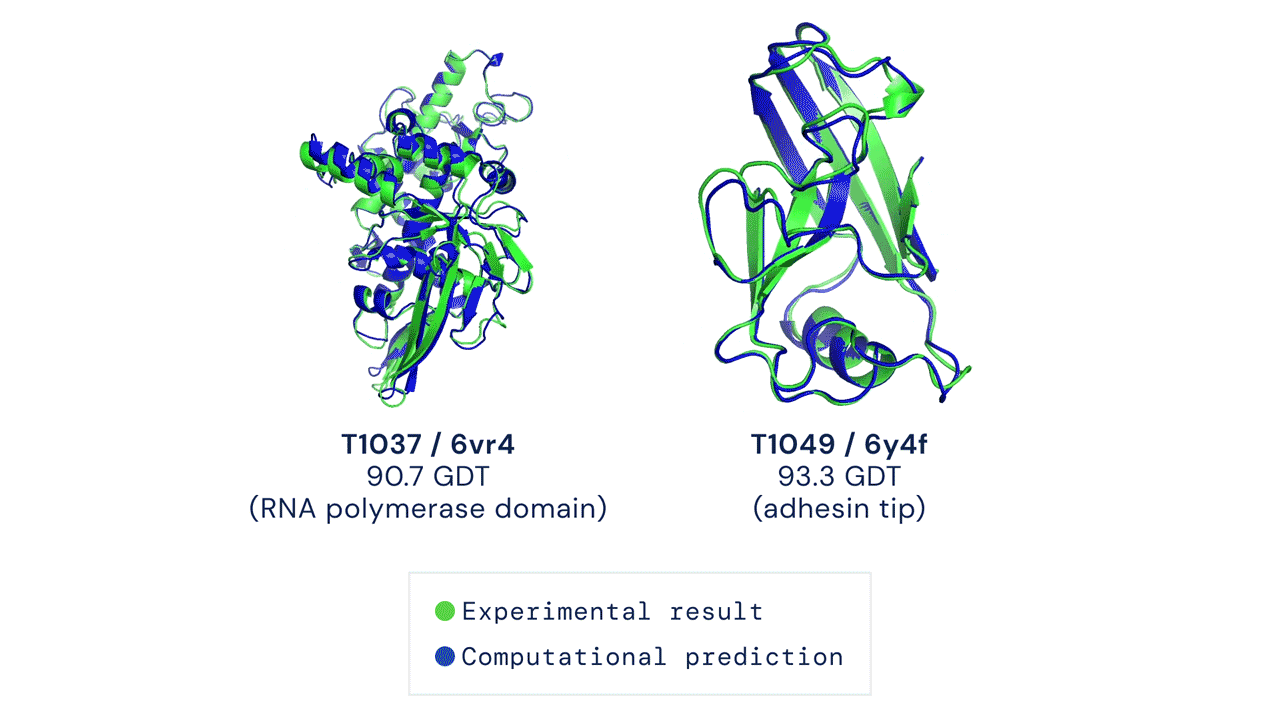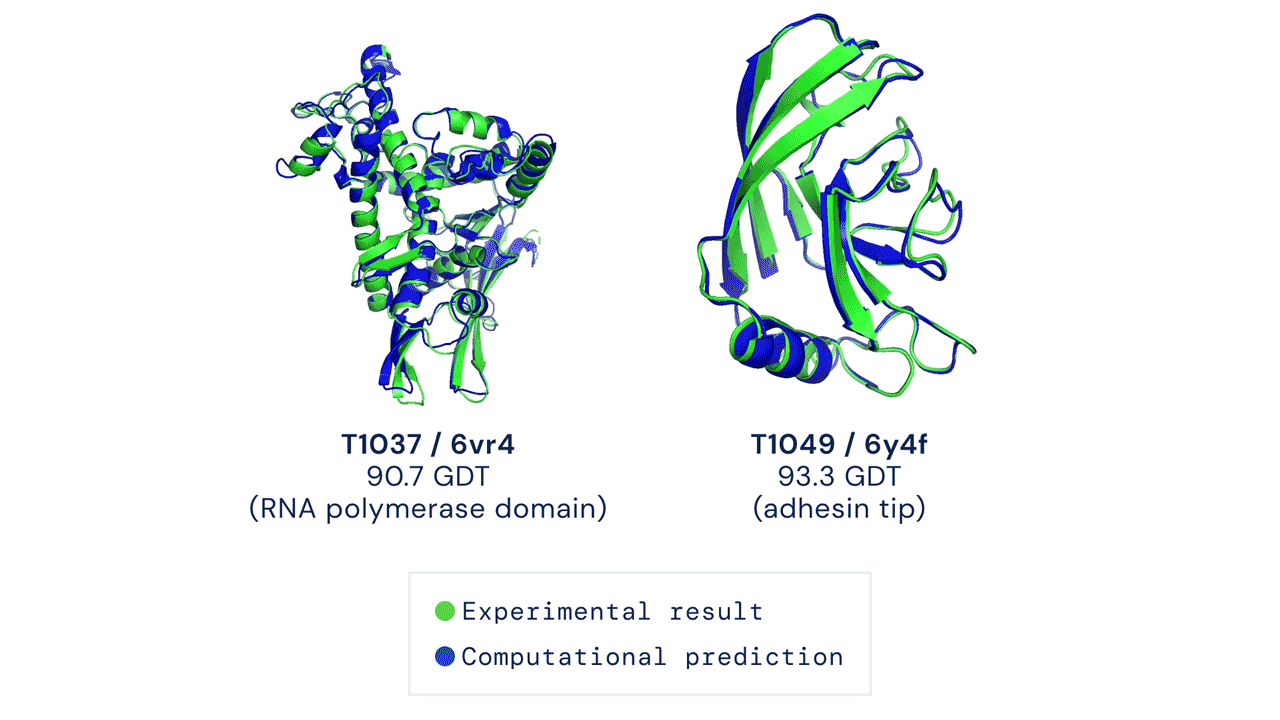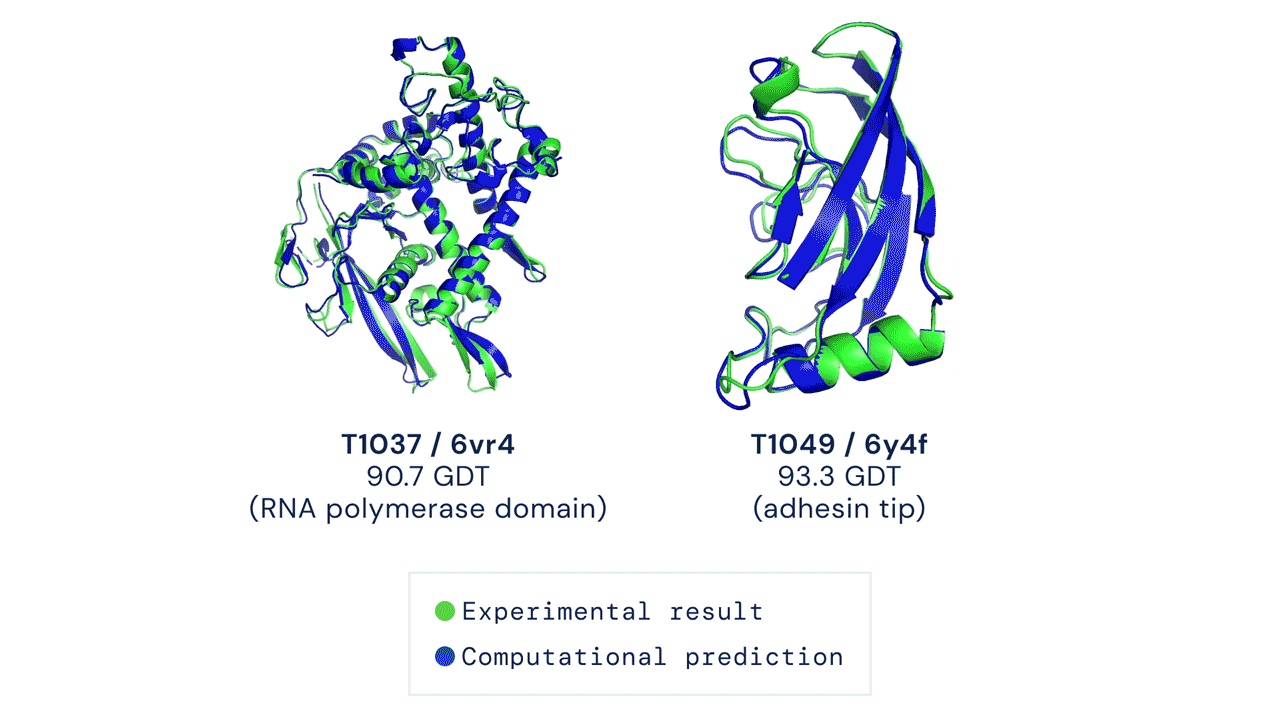
Installation and running your first prediction
You will need a machine running Linux, AlphaFold does not support other
operating systems. Full installation requires up to 3 TB of disk space to keep
genetic databases (SSD storage is recommended) and a modern NVIDIA GPU (GPUs
with more memory can predict larger protein structures).
Please follow these steps:
-
Install Docker.
- Install
NVIDIA Container Toolkit
for GPU support. - Setup running
Docker as a non-root user.
- Install
-
Clone this repository and
cdinto it.bash git clone https://github.com/deepmind/alphafold.git cd ./alphafold -
Download genetic databases and model parameters:
-
Install
aria2c. On most Linux distributions it is available via the
package manager as thearia2package (on Debian-based distributions this
can be installed by runningsudo apt install aria2). -
Please use the script
scripts/download_all_data.shto download
and set up full databases. This may take substantial time (download size is
556 GB), so we recommend running this script in the background:
bash scripts/download_all_data.sh <DOWNLOAD_DIR> > download.log 2> download_all.log &-
Note: The download directory
<DOWNLOAD_DIR>should not be a
subdirectory in the AlphaFold repository directory. If it is, the Docker
build will be slow as the large databases will be copied into the docker
build context. -
It is possible to run AlphaFold with reduced databases; please refer to
the complete documentation.
-
-
Check that AlphaFold will be able to use a GPU by running:
bash docker run --rm --gpus all nvidia/cuda:11.0-base nvidia-smiThe output of this command should show a list of your GPUs. If it doesn't,
check if you followed all steps correctly when setting up the
NVIDIA Container Toolkit
or take a look at the following
NVIDIA Docker issue.If you wish to run AlphaFold using Singularity (a common containerization
platform on HPC systems) we recommend using some of the third party Singularity
setups as linked in https://github.com/deepmind/alphafold/issues/10 or
https://github.com/deepmind/alphafold/issues/24. -
Build the Docker image:
bash docker build -f docker/Dockerfile -t alphafold .If you encounter the following error:
W: GPG error: https://developer.download.nvidia.com/compute/cuda/repos/ubuntu1804/x86_64 InRelease: The following signatures couldn't be verified because the public key is not available: NO_PUBKEY A4B469963BF863CC E: The repository 'https://developer.download.nvidia.com/compute/cuda/repos/ubuntu1804/x86_64 InRelease' is not signed.use the workaround described in
https://github.com/deepmind/alphafold/issues/463#issuecomment-1124881779. -
Install the
run_docker.pydependencies. Note: You may optionally wish to
create a
Python Virtual Environment
to prevent conflicts with your system's Python environment.bash pip3 install -r docker/requirements.txt -
Make sure that the output directory exists (the default is
/tmp/alphafold)
and that you have sufficient permissions to write into it. -
Run
run_docker.pypointing to a FASTA file containing the protein
sequence(s) for which you wish to predict the structure (--fasta_paths
parameter). AlphaFold will search for the available templates before the
date specified by the--max_template_dateparameter; this could be used to
avoid certain templates during modeling.--data_diris the directory with
downloaded genetic databases and--output_diris the absolute path to the
output directory.bash python3 docker/run_docker.py \ --fasta_paths=your_protein.fasta \ --max_template_date=2022-01-01 \ --data_dir=$DOWNLOAD_DIR \ --output_dir=/home/user/absolute_path_to_the_output_dir -
Once the run is over, the output directory shall contain predicted
structures of the target protein. Please check the documentation below for
additional options and troubleshooting tips.
Genetic databases
This step requires aria2c to be installed on your machine.
AlphaFold needs multiple genetic (sequence) databases to run:
- BFD,
- MGnify,
- PDB70,
- PDB (structures in the mmCIF format),
- PDB seqres – only for AlphaFold-Multimer,
- UniRef30 (FKA UniClust30),
- UniProt – only for AlphaFold-Multimer,
- UniRef90.
We provide a script scripts/download_all_data.sh that can be used to download
and set up all of these databases:
-
Recommended default:
bash scripts/download_all_data.sh <DOWNLOAD_DIR>will download the full databases.
-
With
reduced_dbsparameter:bash scripts/download_all_data.sh <DOWNLOAD_DIR> reduced_dbswill download a reduced version of the databases to be used with the
reduced_dbsdatabase preset. This shall be used with the corresponding
AlphaFold parameter--db_preset=reduced_dbslater during the AlphaFold run
(please see AlphaFold parameters section).
📒 Note: The download directory <DOWNLOAD_DIR> should not be a
subdirectory in the AlphaFold repository directory. If it is, the Docker build
will be slow as the large databases will be copied during the image creation.
We don't provide exactly the database versions used in CASP14 – see the
note on reproducibility. Some of the
databases are mirrored for speed, see mirrored databases.
📒 Note: The total download size for the full databases is around 556 GB
and the total size when unzipped is 2.62 TB. Please make sure you have a large
enough hard drive space, bandwidth and time to download. We recommend using an
SSD for better genetic search performance.
📒 Note: If the download directory and datasets don't have full read and
write permissions, it can cause errors with the MSA tools, with opaque
(external) error messages. Please ensure the required permissions are applied,
e.g. with the sudo chmod 755 --recursive "$DOWNLOAD_DIR" command.
The download_all_data.sh script will also download the model parameter files.
Once the script has finished, you should have the following directory structure:
$DOWNLOAD_DIR/ # Total: ~ 2.62 TB (download: 556 GB)
bfd/ # ~ 1.8 TB (download: 271.6 GB)
# 6 files.
mgnify/ # ~ 120 GB (download: 67 GB)
mgy_clusters_2022_05.fa
params/ # ~ 5.3 GB (download: 5.3 GB)
# 5 CASP14 models,
# 5 pTM models,
# 5 AlphaFold-Multimer models,
# LICENSE,
# = 16 files.
pdb70/ # ~ 56 GB (download: 19.5 GB)
# 9 files.
pdb_mmcif/ # ~ 238 GB (download: 43 GB)
mmcif_files/
# About 199,000 .cif files.
obsolete.dat
pdb_seqres/ # ~ 0.2 GB (download: 0.2 GB)
pdb_seqres.txt
small_bfd/ # ~ 17 GB (download: 9.6 GB)
bfd-first_non_consensus_sequences.fasta
uniref30/ # ~ 206 GB (download: 52.5 GB)
# 7 files.
uniprot/ # ~ 105 GB (download: 53 GB)
uniprot.fasta
uniref90/ # ~ 67 GB (download: 34 GB)
uniref90.fasta
bfd/ is only downloaded if you download the full databases, and small_bfd/
is only downloaded if you download the reduced databases.
Model parameters
While the AlphaFold code is licensed under the Apache 2.0 License, the AlphaFold
parameters and CASP15 prediction data are made available under the terms of the
CC BY 4.0 license. Please see the Disclaimer below
for more detail.
The AlphaFold parameters are available from
https://storage.googleapis.com/alphafold/alphafold_params_2022-12-06.tar, and
are downloaded as part of the scripts/download_all_data.sh script. This script
will download parameters for:
- 5 models which were used during CASP14, and were extensively validated for
structure prediction quality (see Jumper et al. 2021, Suppl. Methods 1.12
for details). - 5 pTM models, which were fine-tuned to produce pTM (predicted TM-score) and
(PAE) predicted aligned error values alongside their structure predictions
(see Jumper et al. 2021, Suppl. Methods 1.9.7 for details). - 5 AlphaFold-Multimer models that produce pTM and PAE values alongside their
structure predictions.
Updating existing installation
If you have a previous version you can either reinstall fully from scratch
(remove everything and run the setup from scratch) or you can do an incremental
update that will be significantly faster but will require a bit more work. Make
sure you follow these steps in the exact order they are listed below:
- Update the code.
- Go to the directory with the cloned AlphaFold repository and run
git fetch origin mainto get all code updates.
- Go to the directory with the cloned AlphaFold repository and run
- Update the UniProt, UniRef, MGnify and PDB seqres databases.
- Remove
<DOWNLOAD_DIR>/uniprot. - Run
scripts/download_uniprot.sh <DOWNLOAD_DIR>. - Remove
<DOWNLOAD_DIR>/uniclust30. - Run
scripts/download_uniref30.sh <DOWNLOAD_DIR>. - Remove
<DOWNLOAD_DIR>/uniref90. - Run
scripts/download_uniref90.sh <DOWNLOAD_DIR>. - Remove
<DOWNLOAD_DIR>/mgnify. - Run
scripts/download_mgnify.sh <DOWNLOAD_DIR>. - Remove
<DOWNLOAD_DIR>/pdb_mmcif. It is needed to have PDB SeqRes and
PDB from exactly the same date. Failure to do this step will result in
potential errors when searching for templates when running
AlphaFold-Multimer. - Run
scripts/download_pdb_mmcif.sh <DOWNLOAD_DIR>. - Run
scripts/download_pdb_seqres.sh <DOWNLOAD_DIR>.
- Remove
- Update the model parameters.
- Remove the old model parameters in
<DOWNLOAD_DIR>/params. - Download new model parameters using
scripts/download_alphafold_params.sh <DOWNLOAD_DIR>.
- Remove the old model parameters in
- Follow Running AlphaFold.
Using deprecated model weights
To use the deprecated v2.2.0 AlphaFold-Multimer model weights:
- Change
SOURCE_URLinscripts/download_alphafold_params.shto
https://storage.googleapis.com/alphafold/alphafold_params_2022-03-02.tar,
and download the old parameters. - Change the
_v3to_v2in the multimerMODEL_PRESETSinconfig.py.
To use the deprecated v2.1.0 AlphaFold-Multimer model weights:
- Change
SOURCE_URLinscripts/download_alphafold_params.shto
https://storage.googleapis.com/alphafold/alphafold_params_2022-01-19.tar,
and download the old parameters. - Remove the
_v3in the multimerMODEL_PRESETSinconfig.py.
Running AlphaFold
The simplest way to run AlphaFold is using the provided Docker script. This
was tested on Google Cloud with a machine using the nvidia-gpu-cloud-image
with 12 vCPUs, 85 GB of RAM, a 100 GB boot disk, the databases on an additional
3 TB disk, and an A100 GPU. For your first run, please follow the instructions
from Installation and running your first prediction
section.
-
By default, Alphafold will attempt to use all visible GPU devices. To use a
subset, specify a comma-separated list of GPU UUID(s) or index(es) using the
--gpu_devicesflag. See
GPU enumeration
for more details. -
You can control which AlphaFold model to run by adding the
--model_preset=
flag. We provide the following models:-
monomer: This is the original model used at CASP14 with no
ensembling. -
monomer_casp14: This is the original model used at CASP14 with
num_ensemble=8, matching our CASP14 configuration. This is largely
provided for reproducibility as it is 8x more computationally expensive
for limited accuracy gain (+0.1 average GDT gain on CASP14 domains). -
monomer_ptm: This is the original CASP14 model fine tuned with the
pTM head, providing a pairwise confidence measure. It is slightly less
accurate than the normal monomer model. -
multimer: This is the AlphaFold-Multimer model.
To use this model, provide a multi-sequence FASTA file. In addition, the
UniProt database should have been downloaded.
-
-
You can control MSA speed/quality tradeoff by adding
--db_preset=reduced_dbsor--db_preset=full_dbsto the run command. We
provide the following presets:-
reduced_dbs: This preset is optimized for speed and lower hardware
requirements. It runs with a reduced version of the BFD database. It
requires 8 CPU cores (vCPUs), 8 GB of RAM, and 600 GB of disk space. -
full_dbs: This runs with all genetic databases used at CASP14.
Running the command above with the
monomermodel preset and the
reduced_dbsdata preset would look like this:bash python3 docker/run_docker.py \ --fasta_paths=T1050.fasta \ --max_template_date=2020-05-14 \ --model_preset=monomer \ --db_preset=reduced_dbs \ --data_dir=$DOWNLOAD_DIR \ --output_dir=/home/user/absolute_path_to_the_output_dir -
-
After generating the predicted model, AlphaFold runs a relaxation
step to improve local geometry. By default, only the best model (by
pLDDT) is relaxed (--models_to_relax=best), but also all of the models
(--models_to_relax=all) or none of the models (--models_to_relax=none)
can be relaxed. -
The relaxation step can be run on GPU (faster, but could be less stable) or
CPU (slow, but stable). This can be controlled with--enable_gpu_relax=true
(default) or--enable_gpu_relax=false. -
AlphaFold can re-use MSAs (multiple sequence alignments) for the same
sequence via--use_precomputed_msas=trueoption; this can be useful for
trying different AlphaFold parameters. This option assumes that the
directory structure generated by the first AlphaFold run in the output
directory exists and that the protein sequence is the same.
Running AlphaFold-Multimer
All steps are the same as when running the monomer system, but you will have to
- provide an input fasta with multiple sequences,
- set
--model_preset=multimer,
An example that folds a protein complex multimer.fasta:
python3 docker/run_docker.py \
--fasta_paths=multimer.fasta \
--max_template_date=2020-05-14 \
--model_preset=multimer \
--data_dir=$DOWNLOAD_DIR \
--output_dir=/home/user/absolute_path_to_the_output_dir
By default the multimer system will run 5 seeds per model (25 total predictions)
for a small drop in accuracy you may wish to run a single seed per model. This
can be done via the --num_multimer_predictions_per_model flag, e.g. set it to
--num_multimer_predictions_per_model=1 to run a single seed per model.
AlphaFold prediction speed
The table below reports prediction runtimes for proteins of various lengths. We
only measure unrelaxed structure prediction with three recycles while
excluding runtimes from MSA and template search. When running
docker/run_docker.py with --benchmark=true, this runtime is stored in
timings.json. All runtimes are from a single A100 NVIDIA GPU. Prediction
speed on A100 for smaller structures can be improved by increasing
global_config.subbatch_size in alphafold/model/config.py.
| No. residues | Prediction time (s) |
|---|---|
| 100 | 4.9 |
| 200 | 7.7 |
| 300 | 13 |
| 400 | 18 |
| 500 | 29 |
| 600 | 36 |
| 700 | 53 |
| 800 | 60 |
| 900 | 91 |
| 1,000 | 96 |
| 1,100 | 140 |
| 1,500 | 280 |
| 2,000 | 450 |
| 2,500 | 969 |
| 3,000 | 1,240 |
| 3,500 | 2,465 |
| 4,000 | 5,660 |
| 4,500 | 12,475 |
| 5,000 | 18,824 |
Examples
Below are examples on how to use AlphaFold in different scenarios.
Folding a monomer
Say we have a monomer with the sequence <SEQUENCE>. The input fasta should be:
>sequence_name
<SEQUENCE>
Then run the following command:
python3 docker/run_docker.py \
--fasta_paths=monomer.fasta \
--max_template_date=2021-11-01 \
--model_preset=monomer \
--data_dir=$DOWNLOAD_DIR \
--output_dir=/home/user/absolute_path_to_the_output_dir
Folding a homomer
Say we have a homomer with 3 copies of the same sequence <SEQUENCE>. The input
fasta should be:
>sequence_1
<SEQUENCE>
>sequence_2
<SEQUENCE>
>sequence_3
<SEQUENCE>
Then run the following command:
python3 docker/run_docker.py \
--fasta_paths=homomer.fasta \
--max_template_date=2021-11-01 \
--model_preset=multimer \
--data_dir=$DOWNLOAD_DIR \
--output_dir=/home/user/absolute_path_to_the_output_dir
Folding a heteromer
Say we have an A2B3 heteromer, i.e. with 2 copies of <SEQUENCE A> and 3 copies
of <SEQUENCE B>. The input fasta should be:
>sequence_1
<SEQUENCE A>
>sequence_2
<SEQUENCE A>
>sequence_3
<SEQUENCE B>
>sequence_4
<SEQUENCE B>
>sequence_5
<SEQUENCE B>
Then run the following command:
python3 docker/run_docker.py \
--fasta_paths=heteromer.fasta \
--max_template_date=2021-11-01 \
--model_preset=multimer \
--data_dir=$DOWNLOAD_DIR \
--output_dir=/home/user/absolute_path_to_the_output_dir
Folding multiple monomers one after another
Say we have a two monomers, monomer1.fasta and monomer2.fasta.
We can fold both sequentially by using the following command:
python3 docker/run_docker.py \
--fasta_paths=monomer1.fasta,monomer2.fasta \
--max_template_date=2021-11-01 \
--model_preset=monomer \
--data_dir=$DOWNLOAD_DIR \
--output_dir=/home/user/absolute_path_to_the_output_dir
Folding multiple multimers one after another
Say we have a two multimers, multimer1.fasta and multimer2.fasta.
We can fold both sequentially by using the following command:
python3 docker/run_docker.py \
--fasta_paths=multimer1.fasta,multimer2.fasta \
--max_template_date=2021-11-01 \
--model_preset=multimer \
--data_dir=$DOWNLOAD_DIR \
--output_dir=/home/user/absolute_path_to_the_output_dir
AlphaFold output
The outputs will be saved in a subdirectory of the directory provided via the
--output_dir flag of run_docker.py (defaults to /tmp/alphafold/). The
outputs include the computed MSAs, unrelaxed structures, relaxed structures,
ranked structures, raw model outputs, prediction metadata, and section timings.
The --output_dir directory will have the following structure:
<target_name>/
features.pkl
ranked_{0,1,2,3,4}.pdb
ranking_debug.json
relax_metrics.json
relaxed_model_{1,2,3,4,5}.pdb
result_model_{1,2,3,4,5}.pkl
timings.json
unrelaxed_model_{1,2,3,4,5}.pdb
msas/
bfd_uniref_hits.a3m
mgnify_hits.sto
uniref90_hits.sto
The contents of each output file are as follows:
features.pkl– Apicklefile containing the input feature NumPy arrays
used by the models to produce the structures.unrelaxed_model_*.pdb– A PDB format text file containing the predicted
structure, exactly as outputted by the model.relaxed_model_*.pdb– A PDB format text file containing the predicted
structure, after performing an Amber relaxation procedure on the unrelaxed
structure prediction (see Jumper et al. 2021, Suppl. Methods 1.8.6 for
details).ranked_*.pdb– A PDB format text file containing the predicted structures,
after reordering by model confidence. Hereranked_i.pdbshould contain
the prediction with the (i + 1)-th highest confidence (so that
ranked_0.pdbhas the highest confidence). To rank model confidence, we use
predicted LDDT (pLDDT) scores (see Jumper et al. 2021, Suppl. Methods 1.9.6
for details). If--models_to_relax=allthen all ranked structures are
relaxed. If--models_to_relax=bestthen onlyranked_0.pdbis relaxed
(the rest are unrelaxed). If--models_to_relax=none, then the ranked
structures are all unrelaxed.ranking_debug.json– A JSON format text file containing the pLDDT values
used to perform the model ranking, and a mapping back to the original model
names.relax_metrics.json– A JSON format text file containing relax metrics, for
instance remaining violations.timings.json– A JSON format text file containing the times taken to run
each section of the AlphaFold pipeline.msas/- A directory containing the files describing the various genetic
tool hits that were used to construct the input MSA.-
result_model_*.pkl– Apicklefile containing a nested dictionary of the
various NumPy arrays directly produced by the model. In addition to the
output of the structure module, this includes auxiliary outputs such as:- Distograms (
distogram/logitscontains a NumPy array of shape [N_res,]
N_res, N_bins anddistogram/bin_edgescontains the definition of the
bins). - Per-residue pLDDT scores (
plddtcontains a NumPy array of shape
[N_res] with the range of possible values from0to100, where100
means most confident). This can serve to identify sequence regions
predicted with high confidence or as an overall per-target confidence
score when averaged across residues. - Present only if using pTM models: predicted TM-score (
ptmfield
contains a scalar). As a predictor of a global superposition metric,
this score is designed to also assess whether the model is confident in
the overall domain packing. - Present only if using pTM models: predicted pairwise aligned errors
(predicted_aligned_errorcontains a NumPy array of shape [N_res,]
N_res with the range of possible values from0to
max_predicted_aligned_error, where0means most confident). This can
serve for a visualisation of domain packing confidence within the
structure.
- Distograms (
The pLDDT confidence measure is stored in the B-factor field of the output PDB
files (although unlike a B-factor, higher pLDDT is better, so care must be taken
when using for tasks such as molecular replacement).
This code has been tested to match mean top-1 accuracy on a CASP14 test set with
pLDDT ranking over 5 model predictions (some CASP targets were run with earlier
versions of AlphaFold and some had manual interventions; see our forthcoming
publication for details). Some targets such as T1064 may also have high
individual run variance over random seeds.
Inferencing many proteins
The provided inference script is optimized for predicting the structure of a
single protein, and it will compile the neural network to be specialized to
exactly the size of the sequence, MSA, and templates. For large proteins, the
compile time is a negligible fraction of the runtime, but it may become more
significant for small proteins or if the multi-sequence alignments are already
precomputed. In the bulk inference case, it may make sense to use our
make_fixed_size function to pad the inputs to a uniform size, thereby reducing
the number of compilations required.
We do not provide a bulk inference script, but it should be straightforward to
develop on top of the RunModel.predict method with a parallel system for
precomputing multi-sequence alignments. Alternatively, this script can be run
repeatedly with only moderate overhead.
Note on CASP14 reproducibility
AlphaFold's output for a small number of proteins has high inter-run variance,
and may be affected by changes in the input data. The CASP14 target T1064 is a
notable example; the large number of SARS-CoV-2-related sequences recently
deposited changes its MSA significantly. This variability is somewhat mitigated
by the model selection process; running 5 models and taking the most confident.
To reproduce the results of our CASP14 system as closely as possible you must
use the same database versions we used in CASP. These may not match the default
versions downloaded by our scripts.
For genetics:
- UniRef90:
v2020_01 - MGnify:
v2018_12 - Uniclust30: v2018_08
- BFD: only version available
For templates:
- PDB: (downloaded 2020-05-14)
- PDB70:
2020-05-13
An alternative for templates is to use the latest PDB and PDB70, but pass the
flag --max_template_date=2020-05-14, which restricts templates only to
structures that were available at the start of CASP14.
Citing this work
If you use the code or data in this package, please cite:
@Article{AlphaFold2021,
author = {Jumper, John and Evans, Richard and Pritzel, Alexander and Green, Tim and Figurnov, Michael and Ronneberger, Olaf and Tunyasuvunakool, Kathryn and Bates, Russ and {\v{Z}}{\'\i}dek, Augustin and Potapenko, Anna and Bridgland, Alex and Meyer, Clemens and Kohl, Simon A A and Ballard, Andrew J and Cowie, Andrew and Romera-Paredes, Bernardino and Nikolov, Stanislav and Jain, Rishub and Adler, Jonas and Back, Trevor and Petersen, Stig and Reiman, David and Clancy, Ellen and Zielinski, Michal and Steinegger, Martin and Pacholska, Michalina and Berghammer, Tamas and Bodenstein, Sebastian and Silver, David and Vinyals, Oriol and Senior, Andrew W and Kavukcuoglu, Koray and Kohli, Pushmeet and Hassabis, Demis},
journal = {Nature},
title = {Highly accurate protein structure prediction with {AlphaFold}},
year = {2021},
volume = {596},
number = {7873},
pages = {583--589},
doi = {10.1038/s41586-021-03819-2}
}
In addition, if you use the AlphaFold-Multimer mode, please cite:
@article {AlphaFold-Multimer2021,
author = {Evans, Richard and O{\textquoteright}Neill, Michael and Pritzel, Alexander and Antropova, Natasha and Senior, Andrew and Green, Tim and {\v{Z}}{\'\i}dek, Augustin and Bates, Russ and Blackwell, Sam and Yim, Jason and Ronneberger, Olaf and Bodenstein, Sebastian and Zielinski, Michal and Bridgland, Alex and Potapenko, Anna and Cowie, Andrew and Tunyasuvunakool, Kathryn and Jain, Rishub and Clancy, Ellen and Kohli, Pushmeet and Jumper, John and Hassabis, Demis},
journal = {bioRxiv},
title = {Protein complex prediction with AlphaFold-Multimer},
year = {2021},
elocation-id = {2021.10.04.463034},
doi = {10.1101/2021.10.04.463034},
URL = {https://www.biorxiv.org/content/early/2021/10/04/2021.10.04.463034},
eprint = {https://www.biorxiv.org/content/early/2021/10/04/2021.10.04.463034.full.pdf},
}
Community contributions
Colab notebooks provided by the community (please note that these notebooks may
vary from our full AlphaFold system and we did not validate their accuracy):
- The
ColabFold AlphaFold2 notebook
by Martin Steinegger, Sergey Ovchinnikov and Milot Mirdita, which uses an
API hosted at the Södinglab based on the MMseqs2 server
(Mirdita et al. 2019, Bioinformatics)
for the multiple sequence alignment creation.
Acknowledgements
AlphaFold communicates with and/or references the following separate libraries
and packages:
- Abseil
- Biopython
- Chex
- Colab
- Docker
- HH Suite
- HMMER Suite
- Haiku
- Immutabledict
- JAX
- Kalign
- matplotlib
- ML Collections
- NumPy
- OpenMM
- OpenStructure
- pandas
- pymol3d
- SciPy
- Sonnet
- TensorFlow
- Tree
- tqdm
We thank all their contributors and maintainers!
Get in Touch
If you have any questions not covered in this overview, please contact the
AlphaFold team at alphafold@deepmind.com.
We would love to hear your feedback and understand how AlphaFold has been useful
in your research. Share your stories with us at
alphafold@deepmind.com.
License and Disclaimer
This is not an officially supported Google product.
Copyright 2022 DeepMind Technologies Limited.
AlphaFold Code License
Licensed under the Apache License, Version 2.0 (the "License"); you may not use
this file except in compliance with the License. You may obtain a copy of the
License at https://www.apache.org/licenses/LICENSE-2.0.
Unless required by applicable law or agreed to in writing, software distributed
under the License is distributed on an "AS IS" BASIS, WITHOUT WARRANTIES OR
CONDITIONS OF ANY KIND, either express or implied. See the License for the
specific language governing permissions and limitations under the License.
Model Parameters License
The AlphaFold parameters are made available under the terms of the Creative
Commons Attribution 4.0 International (CC BY 4.0) license. You can find details
at: https://creativecommons.org/licenses/by/4.0/legalcode
Third-party software
Use of the third-party software, libraries or code referred to in the
Acknowledgements section above may be governed by separate
terms and conditions or license provisions. Your use of the third-party
software, libraries or code is subject to any such terms and you should check
that you can comply with any applicable restrictions or terms and conditions
before use.
Mirrored Databases
The following databases have been mirrored by DeepMind, and are available with
reference to the following:
-
BFD (unmodified), by Steinegger M. and Söding J.,
available under a
Creative Commons Attribution-ShareAlike 4.0 International License. -
BFD (modified), by Steinegger M. and Söding J.,
modified by DeepMind, available under a
Creative Commons Attribution-ShareAlike 4.0 International License.
See the Methods section of the
AlphaFold proteome paper
for details. -
Uniref30: v2021_03
(unmodified), by Mirdita M. et al., available under a
Creative Commons Attribution-ShareAlike 4.0 International License. -
MGnify: v2022_05
(unmodified), by Mitchell AL et al., available free of all copyright
restrictions and made fully and freely available for both non-commercial and
commercial use under
CC0 1.0 Universal (CC0 1.0) Public Domain Dedication.